TRD#3: Appion: An enabling tool for EM processing and analysis of complex systems.
Appion was initially developed to enable systematic evaluations of p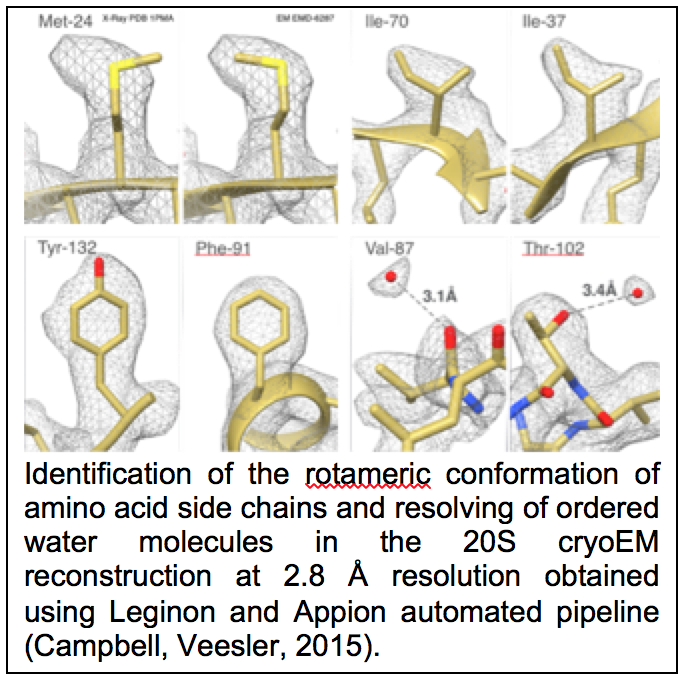 rotocols for image processing and analysis in order to optimize conditions for achieving the highest resolution reconstructions. However, Appion soon became established as a more general enabling technology, providing ready access to a rich variety of disparate structural EM approaches and opening up novel possibilities for investigating structural changes and dynamics in large macromolecular complexes. It also provided a way to readily introduce new members of the community to the methods and offer them a straightforward way to access a wide variety of algorithms and methods using a standard web-based user interface and reporting tools. Our goal in the current funding period was to build on this potential to develop Appion into more mature infrastructure that could be readily distributed, maintained and extended, as well as to develop novel algorithms. Infrastructure development during the last period included major upgrades to the base code that supports web-based image display; building various new base and extended classes to generalize the GUI job launch pages; code development in support of cluster management. Some of the other efforts in this TRD included the following:
rotocols for image processing and analysis in order to optimize conditions for achieving the highest resolution reconstructions. However, Appion soon became established as a more general enabling technology, providing ready access to a rich variety of disparate structural EM approaches and opening up novel possibilities for investigating structural changes and dynamics in large macromolecular complexes. It also provided a way to readily introduce new members of the community to the methods and offer them a straightforward way to access a wide variety of algorithms and methods using a standard web-based user interface and reporting tools. Our goal in the current funding period was to build on this potential to develop Appion into more mature infrastructure that could be readily distributed, maintained and extended, as well as to develop novel algorithms. Infrastructure development during the last period included major upgrades to the base code that supports web-based image display; building various new base and extended classes to generalize the GUI job launch pages; code development in support of cluster management. Some of the other efforts in this TRD included the following:
Maskiton: We developed an interactive, web-based tool, called Maskiton, for creating custom masks and performing 2D classifications on aligned single-particle EM images (Yoshioka, Lyumkis, 2013). The Maskiton interface makes it considerably easier and faster to explore the significance of heterogeneity in single-particle datasets. Driving Biological Projects included the Ltn1 E3 ubiquitin ligase (Lyumkis, Doamekpor, 2013), and assembly intermediates of 30S ribosomal subunits (Sashital, Greeman, 2014).
Optimod: 3D reconstruction and refinement algorithms need an initial model as a starting point. We developed a general, automated procedure, called OptiMod, for reconstructing and optimizing 3D models using common-lines methodologies (Lyumkis, Vinterbo, 2013). OptiMod approximates orientation angles and reconstructs independent maps from 2D class averages. It then iterates the procedure, while considering each map as a raw solution that needs to be compared with other possible outcomes. We incorporated procedures for 3D alignment, clustering, and refinement to optimize each map, as well as standard scoring metrics to facilitate the selection of the optimal model. We showed that small angle tilt-pair data can be included as one of the scoring metrics to improve the selection of the optimal initial model, and also to provide a validation check. The overall approach was validated using two experimental cryo-EM data sets – the 80S ribosome that represents a relatively straightforward case for ab initio reconstruction, and the Tf-TfR complex that represents a challenging case in that it has previously been shown to provide multiple equally plausible solutions to the initial model problem. Optimod is now in regular and routine use as part of the Appion pipeline.
Frealign9: NRAMM personnel worked closely with Niko Grigorieff during his sabbatical year with us to implement a maximum likelihood classification method into the Frealign software (Lyumkis, Brilot, 2013). Particle alignment parameters are determined by maximizing a joint likelihood that can include hierarchical priors, while classification is performed by expectation maximization of a marginal likelihood. The software, Frealign9, was tested using a simulated dataset containing computer-generated projection images of three different 70S ribosome structures, as well as a publicly available dataset of 70S ribosomes. The results showed that the mixed strategy of the new Frealign algorithm yields performance on par with other maximum likelihood implementations, while remaining computationally efficient.
CTF estimation: NRAMM contributed to developing an improved metho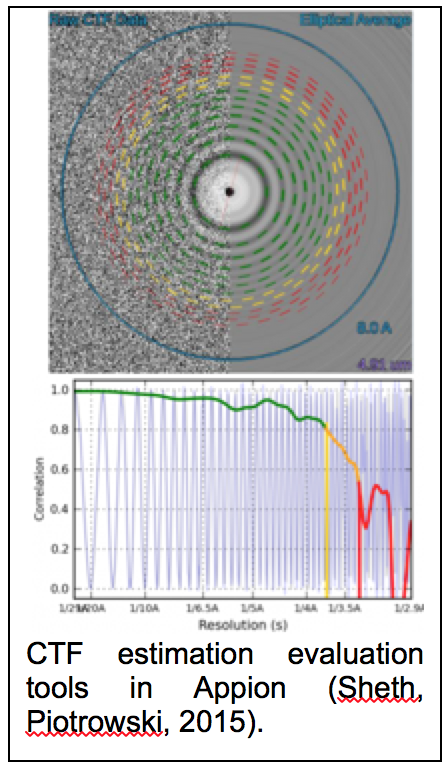 d for extracting the CTF signal from image power spectra and evaluating the quality of the CTF estimation parameters (Sheth, Piotrowski, 2015). This approach removes evaluation range bias and provides a confidence estimate of the CTF estimation (figure 1.9) that allows for direct comparison of multiple estimation runs. This enabled a completely novice user to perform almost as well as the most experienced developers in the CTF Estimation Challenge Event (Marabini, Carragher, 2015). It was also an important aspect in being able to rapidly sort out the highest quality images to achieve the 2.8A map of the proteasome (Campbell, Veesler, 2015). Several additional CTF estimation routines (XMIPP CTF (Sorzano, Jonic, 2007), CTFFIND4 (Rohou and Grigorieff, 2015), gCTF (Zhang, 2016)) were also added as options to the Appion pipeline.
d for extracting the CTF signal from image power spectra and evaluating the quality of the CTF estimation parameters (Sheth, Piotrowski, 2015). This approach removes evaluation range bias and provides a confidence estimate of the CTF estimation (figure 1.9) that allows for direct comparison of multiple estimation runs. This enabled a completely novice user to perform almost as well as the most experienced developers in the CTF Estimation Challenge Event (Marabini, Carragher, 2015). It was also an important aspect in being able to rapidly sort out the highest quality images to achieve the 2.8A map of the proteasome (Campbell, Veesler, 2015). Several additional CTF estimation routines (XMIPP CTF (Sorzano, Jonic, 2007), CTFFIND4 (Rohou and Grigorieff, 2015), gCTF (Zhang, 2016)) were also added as options to the Appion pipeline.
Protomo: Protomo is a software package used in electron tomography for marker-free alignment and 3D reconstruction of tilt series. The marker-free alignment is based on cross-correlation methods and projection matching and this aspect makes the package particularly attractive for in situ cryoelectron tomography. NRAMM worked with Alex Noble and Scott Stagg (FSU) to implement a fully functional Protomo application into Appion (Noble and Stagg, 2015). Protomo has now been added to the Leginon/Appion 3.2 distribution that includes an installer that incorporates Protomo and its dependencies together with the other packages included in Leginon/Appion distribution.
Numerous additional features were also added to Appion including support for a number of options for movie and particle stack frame alignments, several additional 2D alignment and classification algorithms, and options for either Relion or Frelaign9 3D multi-model reconstruction and refinement. A total of ~1600 revisions were made to the Appion base code over the past 4 years.
References
Campbell, M.G., Veesler, D., Cheng, A., Potter, C.S., and Carragher, B. (2015). 2.8 A resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. Elife 4.
Lyumkis, D., Brilot, A.F., Theobald, D.L., and Grigorieff, N. (2013a). Likelihood-based classification of cryo-EM images using FREALIGN. J Struct Biol 183, 377-388.
Lyumkis, D., Doamekpor, S.K., Bengtson, M.H., Lee, J.W., Toro, T.B., Petroski, M.D., Lima, C.D., Potter, C.S., Carragher, B., and Joazeiro, C.A. (2013b). Single-particle EM reveals extensive conformational variability of the Ltn1 E3 ligase. Proc Natl Acad Sci U S A 110, 1702-1707.
Lyumkis, D., Vinterbo, S., Potter, C.S., and Carragher, B. (2013c). Optimod–an automated approach for constructing and optimizing initial models for single-particle electron microscopy. J Struct Biol 184, 417-426.
Marabini, R., Carragher, B., Chen, S., Chen, J., Cheng, A., Downing, K.H., Frank, J., Grassucci, R.A., Bernard Heymann, J., Jiang, W., et al. (2015). CTF Challenge: Result summary. J Struct Biol 190, 348-359.
Noble, A.J., and Stagg, S.M. (2015). Automated batch fiducial-less tilt-series alignment in Appion using Protomo. J Struct Biol 192, 270-278.
Rohou, A., and Grigorieff, N. (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216-221.
Sashital, D.G., Greeman, C.A., Lyumkis, D., Potter, C.S., Carragher, B., and Williamson, J.R. (2014). A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30S subunit assembly in E. coli. Elife 3.
Sheth, L.K., Piotrowski, A.L., and Voss, N.R. (2015). Visualization and quality assessment of the contrast transfer function estimation. J Struct Biol.
Sorzano, C.O., Jonic, S., Nunez-Ramirez, R., Boisset, N., and Carazo, J.M. (2007). Fast, robust, and accurate determination of transmission electron microscopy contrast transfer function. J Struct Biol 160, 249-262.
Yoshioka, C., Lyumkis, D., Carragher, B., and Potter, C.S. (2013). Maskiton: Interactive, web-based classification of single-particle electron microscopy images. J Struct Biol 182, 155-163.
Zhang, K. (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1-12.